Quickstart Examples
The following two examples may familiarize you with the use of LModeA-nano.
Ethane calculated by Gaussian 16
In the LModeA-nano-main/quickstart/ethane directory, a formatted checkpoint file ethane-pbe.fchk was generated by the vibrational analysis (freq) calculation in Gaussian 16 package.
For this type of input data file, it can be directly loaded to LModeA-nano.
Following these steps
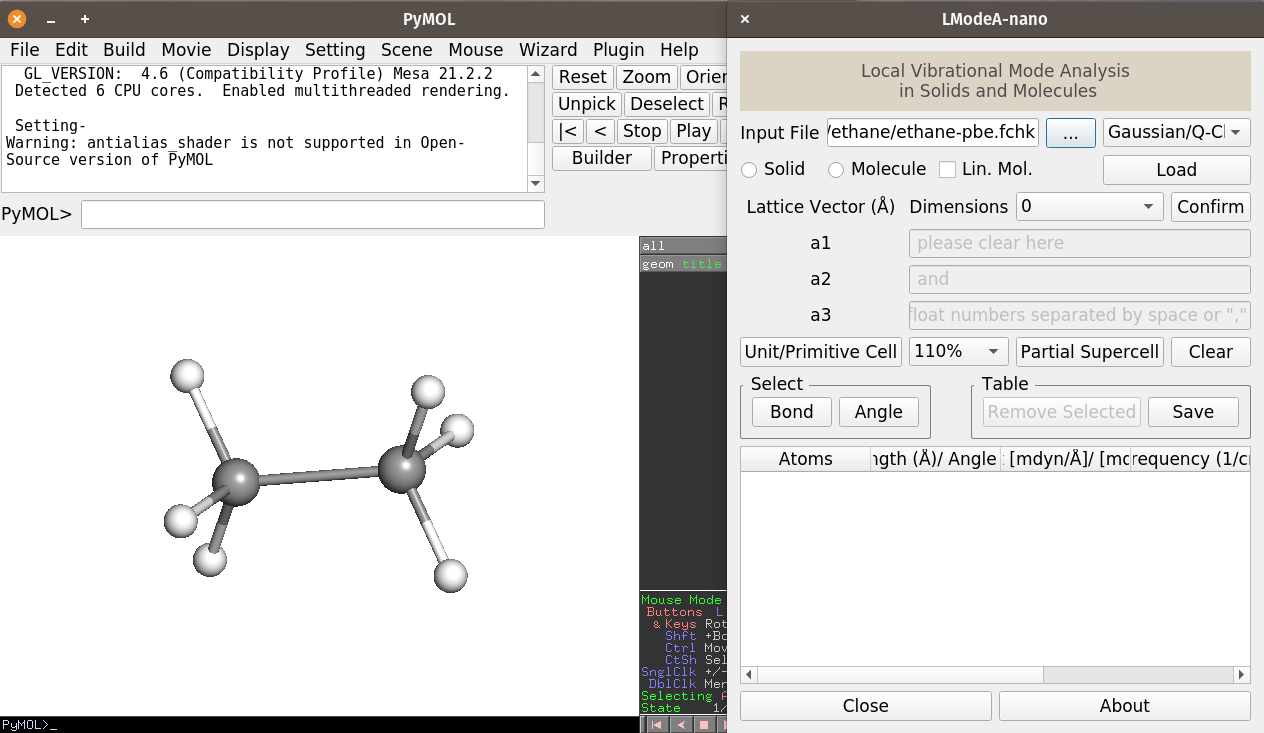
open a new PyMOL window and launch LModeA-nano by clicking Plugin → LModeA-nano
click the … (Browse) button and select the
ethane-pbe.fchkfilechange the program drop-down list from VASP to Gaussian/Q-Chem
click Load
change Dimensions to
0and click Confirm button
We will see the following
By selecting the CC bond for local mode analysis, the result is

2D Graphene calculated by CRYSTAL17
In the LModeA-nano-main/quickstart/graphene directory, two files were generated by the vibrational frequency calculation with CRYSTAL17 program
crystal_21991013.out- CRYSTAL output file of vibrational analysisHESSFREQ.DAT- Hessian file generated after vibrational analysis calculation
In the same directory, we compose an input file crystal.inp for LModeA-nano
@crystal
OUTPUT = crystal_21991013.out
HESSFREQ = HESSFREQ.DAT
Following these steps
open a new PyMOL window and launch LModeA-nano by clicking Plugin → LModeA-nano
click the … (Browse) button and select the
crystal.inpfilechange the program drop-down list from VASP to CRYSTAL
click Load
change Dimensions to
2and click Confirm buttonchange the supercell size percentage from
110%to140%and click Partial Supercell button
We will see the structure like the following
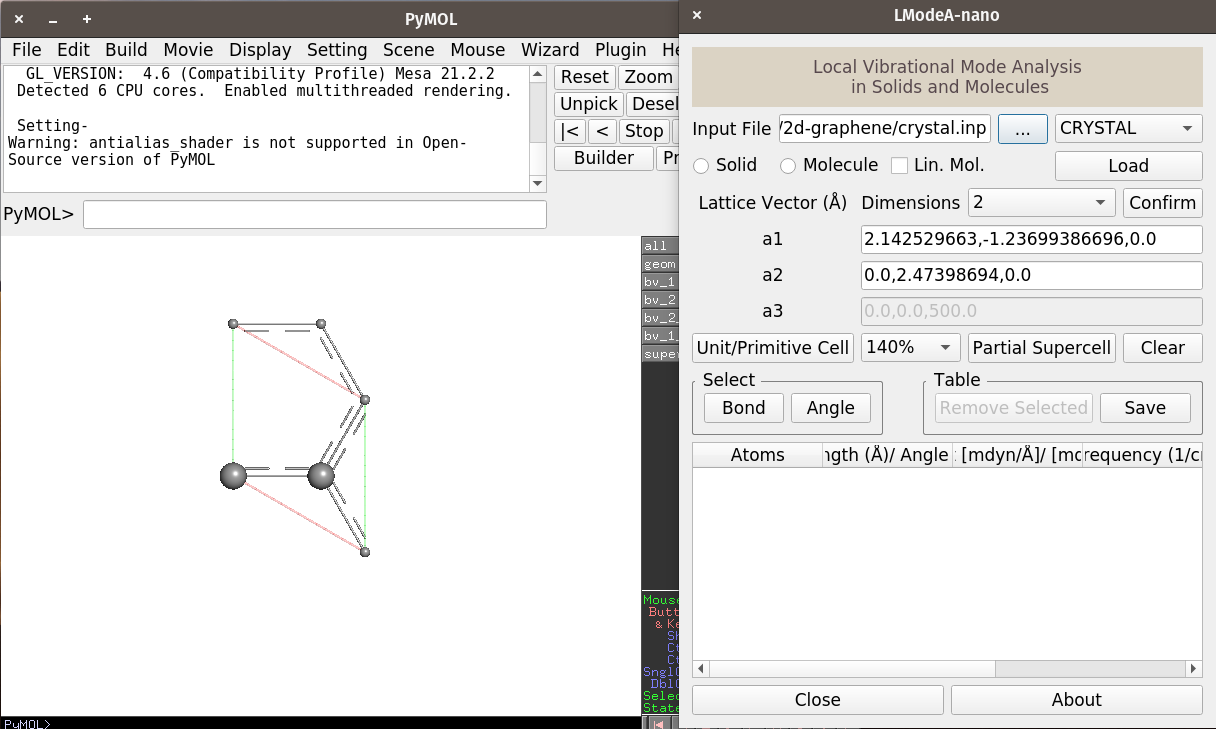
The PyMOL window shows the graphene primitive cell having two carbon atoms (with larger radii) and other carbon atoms belong to neighboring unit cells.
To do local vibrational mode analysis of CC bonds in this structure, click the Bond button in the LModeA-nano window. The interactive wizard may ask for the selection of the first atom of a bond
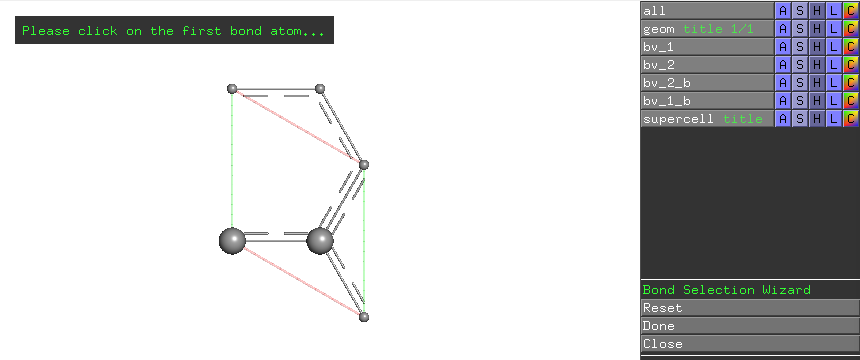
Click the left carbon atom in the primitive cell, the wizard prompts the selection of the second atom of a bond
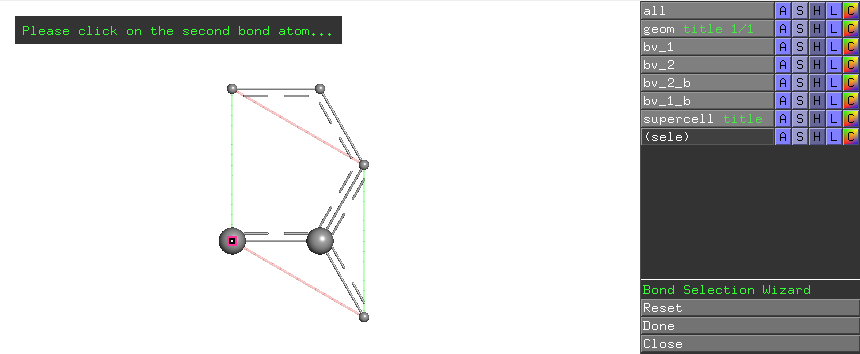
Click the right carbon atom in the primitive cell, the wizard prompts clicking Done button in the wizard menu on the right-hand-side.
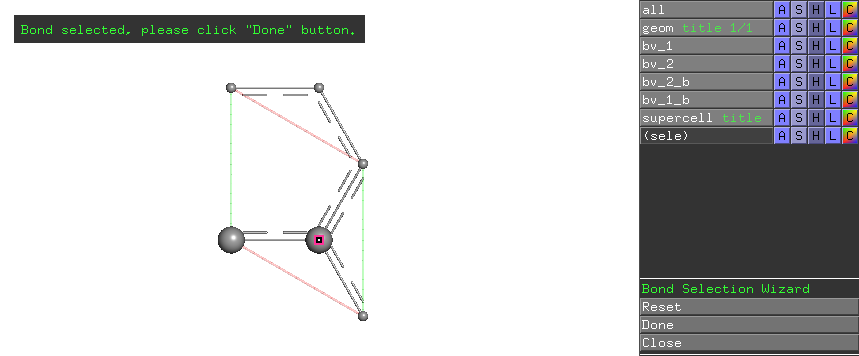
Afterwards, the local mode analysis result for this CC bond is shown in the table region of the LModeA-nano GUI window.

Users are encouraged to try to select a different CC bond in this example for local mode analysis, the result is expected to be the same as for C1-C2 due to symmetry in graphene structure.